脊髓性肌肉萎縮症基因檢測-SMN基因 (SMA)
服務介紹
脊髓性肌肉萎縮症(Spinal Muscular Atrophy,SMA),是種會致命的遺傳疾病,在台灣每40人就有一位是帶因者,是帶因率僅次於海洋性貧血的遺傳疾病。 脊髓性肌肉萎縮症是自體隱性遺傳的疾病,帶因者雖然不會發病,但當夫妻雙方都是帶因者時,胎兒就有 1/4 機會罹患重症。慧智基因提供《脊髓性肌肉萎縮症基因篩檢》,建議孕前或懷孕初期即安排檢查,及後續必要的遺傳諮詢。
■ 脊髓性肌肉萎縮症
脊髓性肌肉萎縮症 (Spinal muscular atrophy, SMA)是一種可以致命的遺傳疾病,發病年齡從出生到成年皆有可能發生。當發病時,患者的肌肉會產生對稱性、逐漸性地退化且軟弱無力的萎縮表現,逐漸影響患者控制隨意肌肉的能力,如走路、爬行、吞嚥、呼吸和控制頭、頸肌肉等日常動作。一般來說,脊髓性肌肉萎縮症依其發病年齡、疾病嚴重度及肌肉受影響程度分為三型。

■ 脊髓性肌肉萎縮症(SMA)遺傳模式
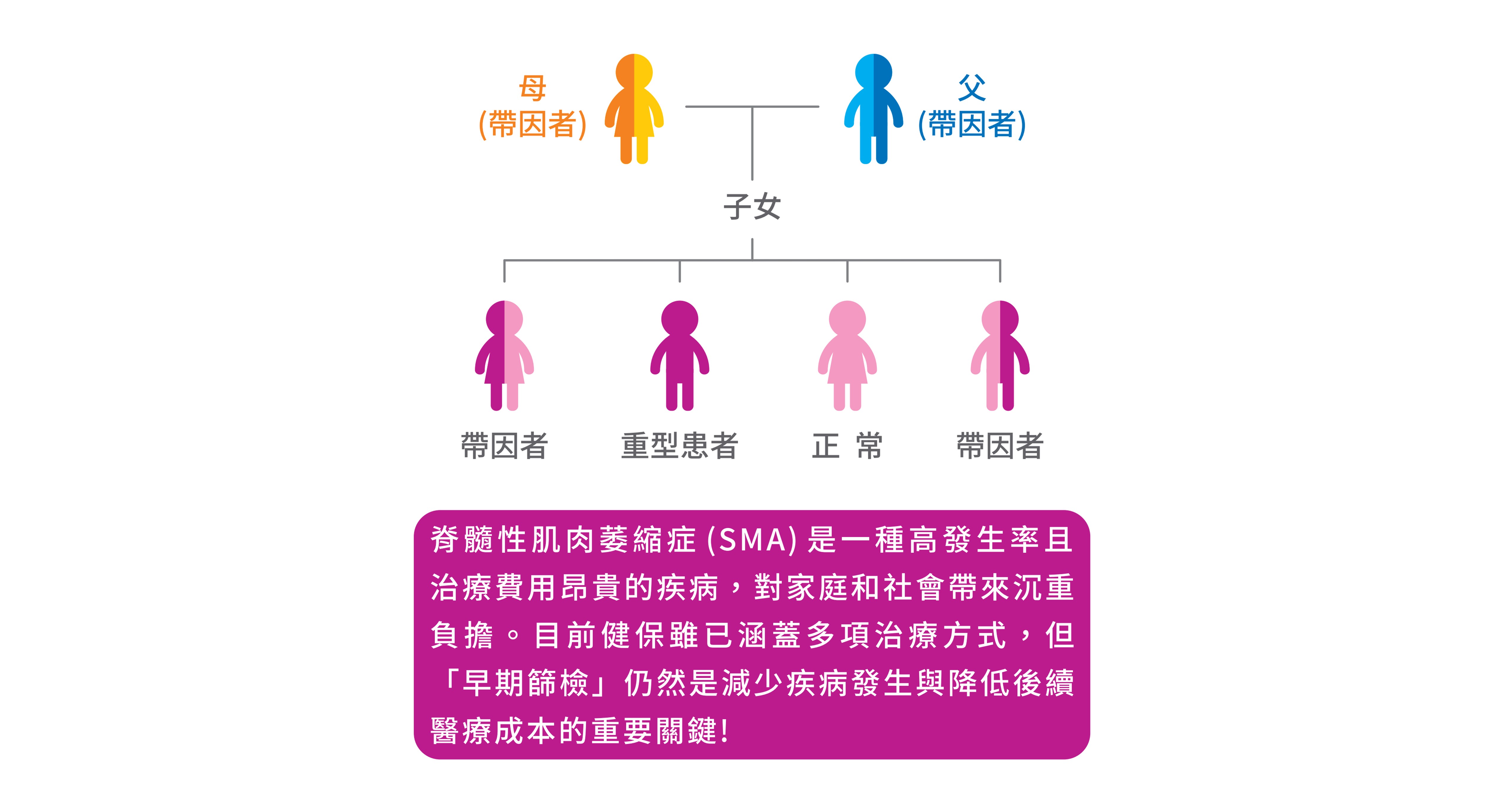
脊髓性肌肉萎縮症的發生主要是因為第五條染色體上一種稱為「運動神經元存活基因」(SMN)產生突變所致。此疾病是自體隱性遺傳的疾病,帶因者雖不會發病,但當父母雙方都是帶因者時,每一胎有四分之一的機會生下罹患重症的寶寶,約95%的脊髓肌肉萎縮症患者是因為SMN1基因出現大片段缺失或轉換導致,其它少數若無SMN1基因大片段缺失或轉換的患者, 則可能是在SMN1基因上發生一些小突變而致病。大部份正常人具有二個以上之SMN1基因,帶因者只具有一個SMN1基因,而病人則完全沒有正常的SMN1基因。台灣約每40人就有一位是帶因者,是帶因率僅次於海洋性貧血的遺傳疾病。
檢測說明
■ 檢測平台
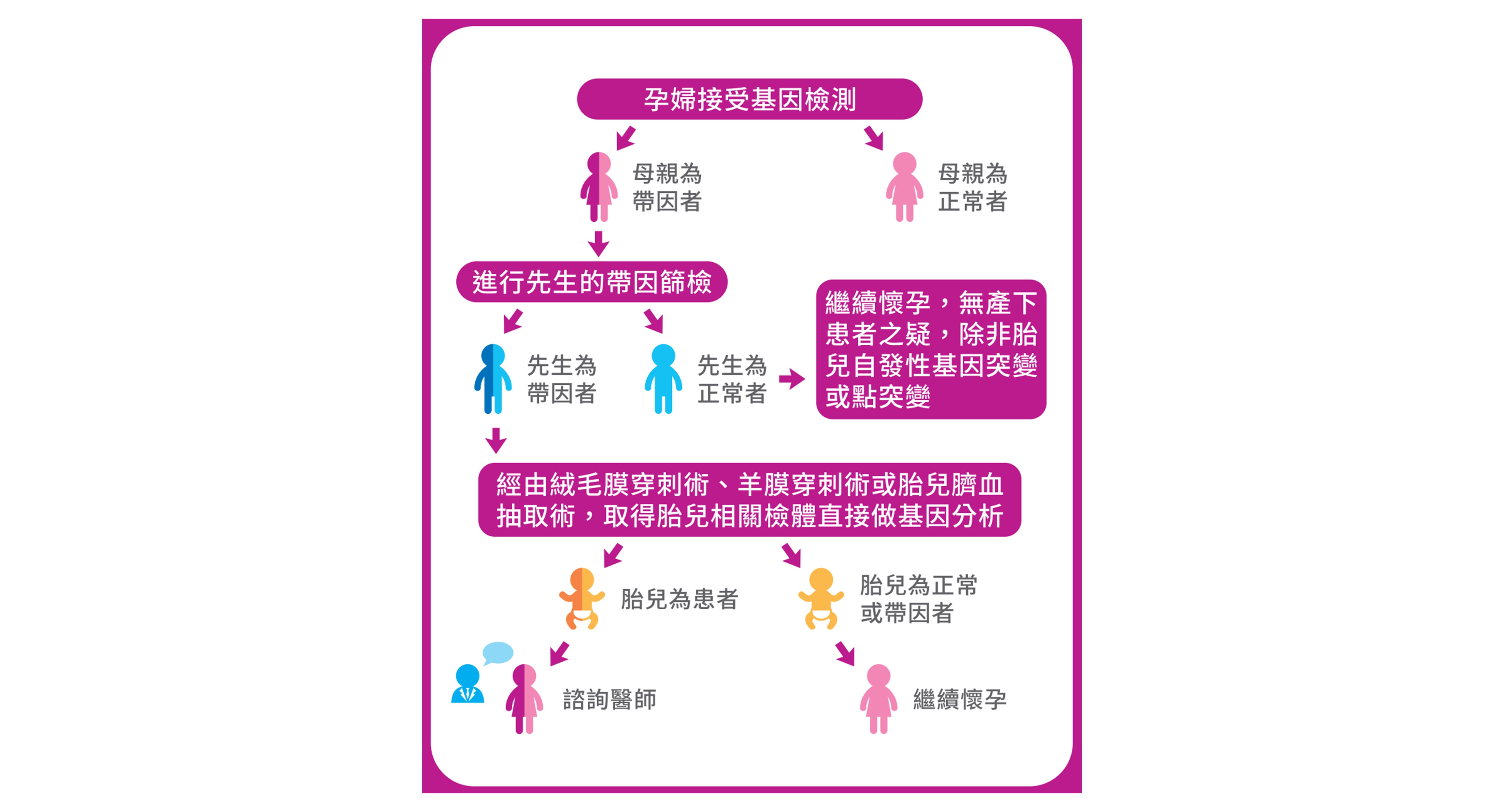
我們自2003年開始研發脊髓性肌肉萎縮症基因檢測之後,終於成功的建立了世界上第一套的脊髓性肌肉萎縮症基因檢測系統,已有18萬人接受過此一檢測,為目前台灣在此一檢測中的領先團隊。此外,在這中間不斷研究與精進測試,目前我們的平台檢測系統(MLPA),可以同時具備高通量、高敏感度與高特異性,通過這樣的高效率篩檢系統,可快速且準確地篩檢出帶因者(準確度達95%~98%),讓處於高危險之家庭,提早於產前清楚知道潛在面臨的風險,並加以因應。
適用對象
✔ 初次篩檢出夫妻皆為脊髓性肌肉萎縮症帶因者
✔ 曾生育過脊髓性肌肉萎縮症患者之夫妻
檢測流程