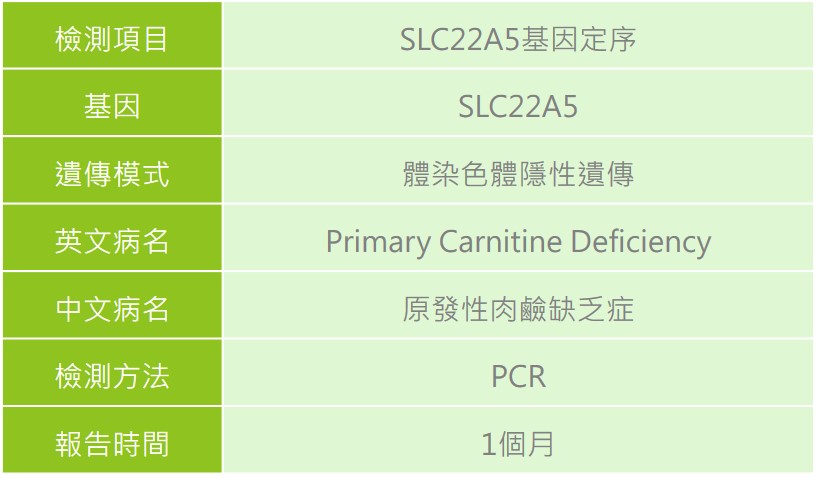
原發性肉鹼缺乏症疾病介紹與衛教
疾病簡介
原發性肉鹼缺乏症(Primary carnitine deficiency, PCD),致病原因為位於第5號染色體上的SLC22A5基因突變所致,該基因突變造成患者缺乏細胞膜上的肉鹼運輸裝置 OCTN2 (organic cation/carnitine transporter 2),導致肉鹼由尿中流失,造成體內肉鹼缺乏,使得無法有效利用脂肪酸來產生能量,也無法產生足夠的酮體為大腦使用,導致可能出現心肌、腦神經、肌肉骨骼等方面出現病變,屬於脂肪酸氧化異常疾病。
臨床症狀
患者臨床表現變異性大,部分患者無任何臨床症狀,有些患者則於新生兒或是幼兒期會發生心肌、中樞神經系統、肌肉骨骼等系統之病變,包括嚴重腦部障礙(腦神經病變)、心臟衰竭及腫大、精神錯亂、肌肉無力、低血糖等臨床症狀,患者易有昏迷或猝死的風險。
患者發病年齡通常介於1歲到7歲(平均好發年齡為2歲)
● 於新生兒期發病,以低酮體性低血糖(Ketotic hypoglycemia)的腦神經病變症狀為主,嚴重可能會導致昏迷猝死的情形;若未能及時的給予肉鹼補充,會出現反覆性的腦神經病變,導致患童出現發展遲緩及中樞神經功能失常的情形,另外也會合併肝腫大、肝臟指數異常及高血氨症。
● 於兒童期發病,以肌肉骨骼及心肌病變較為常見。若未及時診斷補充肉鹼,隨著時間增長會逐漸演變為心衰竭。此外,患者會有腸胃蠕動不良(反覆腹痛及腹瀉)、貧血及反覆性感染等症狀。
患者需終生補充肉鹼、避免飢餓及禁食,也需要定期監控游離肉鹼濃度、心臟及發育生長;若在器官尚未受損前治療與追蹤,通常患者健康狀況良好與正常人無異。
遺傳模式
體染色體隱性遺傳,若父母親雙方皆為同基因突變之帶因者,其本身並不會發病或有任何症狀,則下一代不分性別有 1/4 的機率生下患者。
檢驗方式
在新生兒篩檢異常或醫師進行臨床症狀評估後會進行一系列檢測,實驗室生化檢測項目(檢驗患者血液及尿液中的肉鹼濃度、血氨、血糖、尿酮、肝功能、電解質、尿酸及肌肝酸等數值),並進行心臟超音波、X光等影像學檢查,確認是否發生心臟肥大的情形。另外也會檢測皮膚纖維母細胞中是否缺乏肉鹼運輸裝置(carnitine transporter)、肉鹼停藥測試或是透過分子基因檢測作為診斷。患者及早診斷與補充肉鹼,可降低腦部、心臟及骨骼肌肉等器官發生病變的風險。
首選慧智 (相關檢測如需進一步諮詢,請洽詢專業醫療人員或醫師)
慧智帶因篩檢v2.0/v3.0
https://www.sofivagenomics.com.tw/zh-tw/Product/1/2/item/98
慧智新生兒基因檢測v2.0/v3.0
https://www.sofivagenomics.com.tw/zh-tw/Product/5/3/item/75
罕見疾病基因檢測
https://www.sofivagenomics.com.tw/zh-tw/Product/4/6/item/29
參考資料
● GeneReviews®
● 罕見遺傳疾病一點通
● 財團法人罕見疾病基金會